Below is an overview of some of our research projects.
Characterization of Biomolecular Thermochemistry by Infrared Action Spectroscopy
We are developing novel methods to characterize the conformational landscape of biomolecular ions through quantitative measurement of conformer thermochemistry. A Van ‘t Hoff analysis provides a straightforward approach to measure the relative entropy and enthalpy of conformers in a system at equilibrium. For the analysis of molecular ions in vacuum, the temperature of an ion population can be controlled by varying the temperature of ion-trap buffer gas with which the ions are equilibrated. Temperature-dependent spectroscopic changes can be mapped to changes in conformer population, and construction of a Van ‘t Hoff plot then provides the relative enthalpy and entropy of the populated conformers. However, in standard ion action spectroscopy experiments, this method is limited by the onset of spectral broadening at elevated temperatures, which impedes the assignment of spectral lines and complicates quantification. Infrared action spectroscopy of ions trapped in helium nanodroplets offers an appealing solution to this challenge. Because the rate of dopant cooling following capture is typically faster than the rate of molecular isomerization, entrained molecules can be kinetically trapped in local conformational minima. As a result, spectroscopic characterization of a beam of molecule-doped nanodroplets provides a snapshot of the equilibrium conformer population prior to capture. This unique property enables the study of temperature-dependent conformational equilibria via infrared spectroscopy without the deleterious effects of spectral broadening at higher temperatures, as previously shown for neutral species by Vilesov and co-workers and Douberly and co-workers.
![Figure 2. Helium nanodroplet infrared action spectroscopy of the dinucleotide anion [dTA−H]− for the experimental determination of conformational thermochemistry. Shown in (a) are IR spectra collected with varying ion temperature prior to pickup. Component spectra identified by non-negative matrix factorization (NMF) are shown in (b), and computed harmonic spectra of the low-energy structures (panel d) are shown in (c). A Van ‘t Hoff plot constructed from the population of each component at a given temperature is shown in (e). Adapted from https://doi.org/10.1039/D0CP02482A](https://www.chm.uri.edu/wp4/wp-content/uploads/userfileuploads/dthomas/Thermochem_Summary-01.jpg)
The results from a proof-of-principle study using this approach are shown in Figure 2. A DNA dinucleotide anion, [dTA−H]− (structures shown in Figure 2d), was chosen based upon previous studies suggesting a two-state system well-suited for initial experiments. Shown in Figure 2a are representative IR spectra of ions captured in helium nanodroplets at pre-pickup ion temperatures between 93 K and 342 K. The infrared spectra show a strong dependence on pre-pickup ion temperature, consistent with the preservation of conformer populations upon cooling to 0.4 K within the nanodroplet. Spectral deconvolution using non-negative matrix factorization (NMF) was employed to obtain component spectra and conformer populations at each temperature (Figure 2b). Comparison of the experimental thermochemical parameters (Figure 2e) and NMF spectra to those from theory (Figure 2c, d) enabled an assignment of populated conformers.
We are currently working to expand upon these initial results and preform a more comprehensive assessment of the advantages and limitations of this approach. Ultimately, we envision this method will facilitate the study of “shallow” free energy landscapes, which often exhibit highly dynamic conformer populations in the condensed phase. Examples of such systems include intrinsically disordered regions of peptides and proteins, which are important in cellular regulation processes and also contribute to protein misfolding diseases, and peptide macrocycles, which are currently of great interest as therapeutics for traditionally “undruggable” targets.
This work is supported by the NSF under grant CHE-2212926.
Structural Characterization of Deep Eutectic Solvents
Deep eutectic solvents (DESs) are a rapidly developing class of solvents with tunable solvation properties modulated by the selection of ionic hydrogen bond acceptors (typically quaternary ammonium species) and hydrogen bond donors. These solvents exhibit many of the desirable properties of standard ionic liquids, including low vapor pressure, high thermal stability, and tunable solvation properties. In addition, they possess several favorable attributes distinct from those of ionic liquids such as decreased toxicity, low cost, and comparatively simple preparation. For these reasons, there is great interest in the development of new DESs as well as the investigation of the structure-function relationship across solvent compositions.
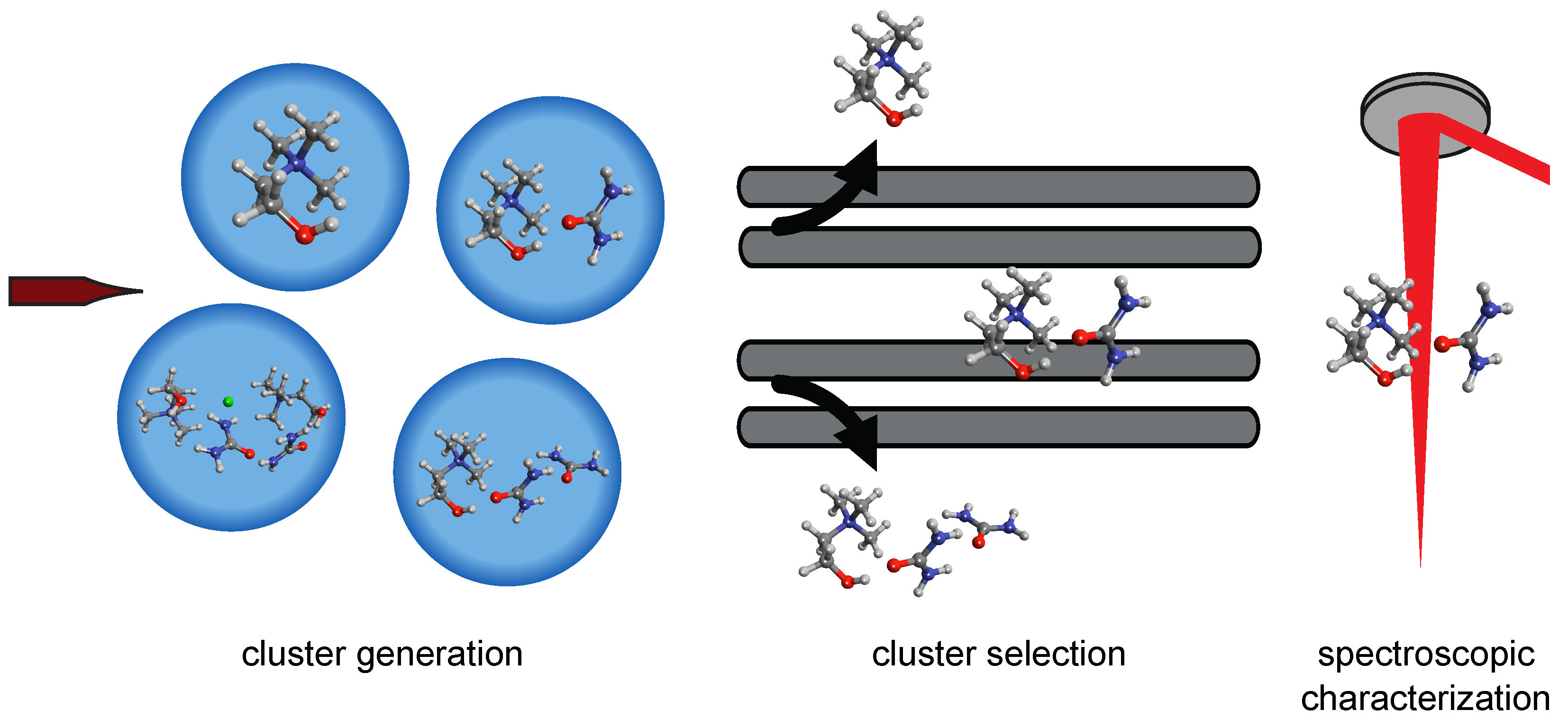
The expansive applications of DESs range from metal processing to the analysis of biomolecular folding and stability. DESs have also shown great promise for industrial chemical processes, particularly as solvents for liquid/liquid extractions. Central to the choice of appropriate DES components is the identification of the underlying structural motifs that govern the observed properties. Although general structural properties have been identified for the most common DES formulations, there remains significant uncertainty in the prevalence and structural contributions of specific interactions, most notably in the hydrogen bonding interactions of the hydrogen bond donors with charged moieties and with one another. Therefore, there exists the need for additional characterization of DES systems to unravel the fundamental intra- and intermolecular interactions prominent in various DES formulations.
We are using a novel approach to study interactions in DESs through the characterization of charged cluster species by infrared photodissociation (IRPD) action spectroscopy and helium nanodroplet isolation infrared (HENDI IR) action spectroscopy using the instrumentation shown in Figure 1. As illustrated in Figure 3, the electrospray process generates a variety of cluster molecules (Figure 3a), with the most intense signal typically arising from the monomeric cation or anion. However, when the instrument is tuned to minimize collisional activation during ion transfer from atmospheric pressure to vacuum, cluster ions are also routinely observed. These clusters are then isolated utilizing the quadrupole mass filter (Figure 3b) and accumulated in the off-axis ion trap. Once trapped, the ions can be characterized by infrared photodissociation (IRPD) spectroscopy or captured by a helium nanodroplet for HENDI IR spectroscopy experiments (Figure 3c). The obtained IR spectra are matched to theoretical spectra of candidate structures computed using electronic structure methods to identify the observed structural motifs. In addition, the IR action spectra of clusters varying in net charge, composition, and size are compared to condensed-phase IR spectra of the relevant DES formulation, thereby enabling an assessment of the connection between cluster structure and the prominent structural motifs in solution. This approach will provide a detailed assessment of potential binding motifs between DES components, providing new insight into DES structure at the nanoscale.
This work is supported by a Doctoral New Investigator grant from the Petroleum Research Fund of the American Chemical Society.
Spectroscopic Investigation of Intermediates in Catalysis
Carbon dioxide is a potentially abundant and accessible carbon source to produce synthetic building blocks as well as chemical fuels. To effectively use this carbon source, however, it is necessary to develop efficient and robust catalysts that can reduce carbon dioxide to more valuable forms such as carbon monoxide, methane, and methanol. The reductive hydrogenation of carbon dioxide to methanol represents a central step in the production of value-added chemicals and also yields a possible storage medium for hydrogen. Therefore, the development of efficient catalysts for this reaction, especially those based on abundant first-row transition metals, is an extremely active area of research. Catalysts that function via a similar reductive hydrogenation mechanism to convert carboxylic acids to alcohols are also highly desirable, as they can be employed in the production of chemical fuels from biomass.
Characterization of the intermediate species in the catalytic cycle is necessary to improve the design and function of catalysts. However, as many species are short-lived, identifying intermediate structures represents a significant experimental challenge. As an alternative to direct characterization in solution, putative reaction intermediates can be generated within a mass spectrometer using a combination of dissociative, ion-ion, and ion-molecule reactions. Although unstable in the condensed phase, reactive intermediates generated in the gas phase can readily be isolated, trapped, and characterized by infrared action spectroscopy. Recent studies have demonstrated the utility of this approach for the study of catalytic reaction cycles, for example in elucidating the binding mode of analytes in CO2 reduction or water oxidation catalysts.
Our laboratory is developing methods to study key intermediates in homogeneous catalysis, with a primary focus on first-row transition metal catalysts used for the reductive hydrogenation of CO2 or carboxylic acids. Key intermediates proposed for many of these catalysts involve the binding of molecular hydrogen to the metal center and subsequent hydride transfer to the target analyte. One such intermediate structure for a carboxylic acid hydrogenation catalyst is shown schematically in Figure 4a. We are working to generate precursors to such intermediate states via electrospray ionization, modify the molecular ions by collision-induced dissociation (to remove ligands), and finally adduct the putative intermediate species with H2 or CO2 within an ion trap or within the helium nanodroplet.
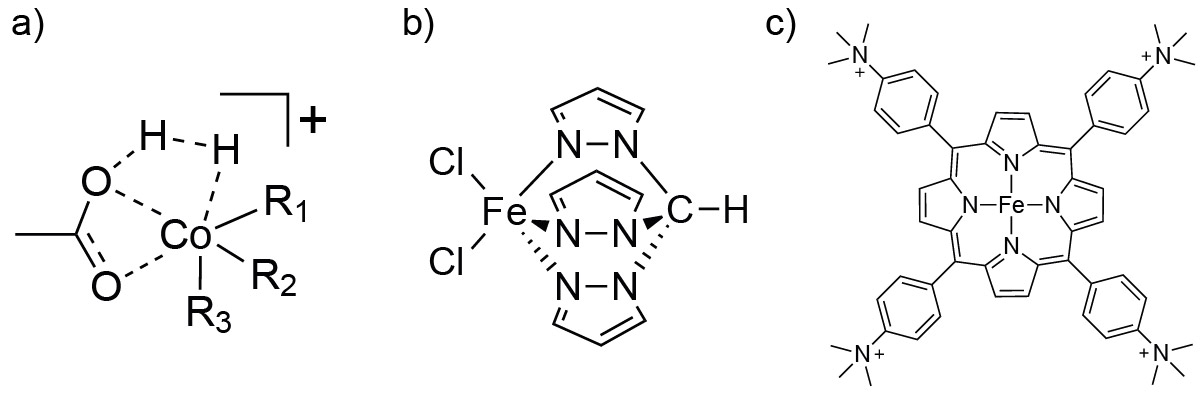
We are further interested in the characterization of systems for which the catalytic mechanism has yet to be proposed, such as the iron scorpionate catalyst for reductive hydrogenation shown in Figure 4b. Additionally, we are interested in investigating species such as the iron porphyrin catalyst shown in Figure 4c, which can reduce CO2 to CO and can also function in the catalytic conversion of CO to methane. A more comprehensive understanding of the intermediate species in the cycle of these catalysts will enable improved ligand selection and catalyst design for better stability and efficiency.
This is an ongoing project, and we are currently working towards collecting preliminary data to support funding applications.
Reactive Species and Ions at Liquid/Vapor Interfaces
The water/air interface is a ubiquitous environment that impacts chemistry throughout the troposphere, including at the ocean surface and in sea spray or fog aerosol. Because of its unique attributes, this transition region frequently serves as a medium for processes unobserved in bulk phases. For example, amphiphilic or highly polarizable analytes are accumulated at the interface, and heterogeneous reactions between these enriched species and gas-phase oxidants can strongly influence the chemical evolution of both the condensed and gas phases. Importantly, the distinctive structure of the water network in this region can stabilize intermediates or transition states, thereby altering the reaction rate or even the underlying reaction mechanism. Reactions where such effects have been invoked include the reaction of chloride ions with hydroxyl radicals (OH), the reaction of ozone with bromide and iodide ions, the reaction of ozone with iron complexes, and the reaction of OH with benzoate ions. These processes can influence air quality by modulating the oxidizing capacity (or “cleaning ability”) of the atmosphere and altering the processing of condensed organics in aerosol, and a better understanding of such reactions is necessary to resolve inconsistencies between experiment and atmospheric models.
Molecular-level insight into these interfacial processes is challenging to achieve, as most experimental methods cannot differentiate between bulk and surface processes or cannot achieve sufficient time resolution to capture these rapid reactions. However, the fine control over reaction conditions afforded by the IR action spectroscopy approach makes this methodology ideal for unraveling the relationship between solvation structure and chemical reactivity at the water/air interface. Utilizing this instrumentation, solvent-ion nanoclusters containing a known number of water molecules can be prepared and complexed with highly reactive gas-phase species such as O3 or OH. These complexes can then be characterized by infrared action spectroscopy to reveal the molecular structure and how it evolves with the size of the nanocluster and the geometry of the water network.
We are especially interested in studying interfacial reactions between halide ions and ozone or hydroxyl radicals. The reaction of Cl− with OH at the liquid/vapor interface of marine sea-salt aerosols has been proposed as an important source of gas-phase Cl2, but there is still a degree of controversy as to the mechanism of this reaction, as the predicted structure of the solvated complex (Figure 5a) is highly sensitive to the level of theory employed. Similarly, the interfacial reaction of Br− with O3 at the liquid/vapor interface is expected to be an important source of precursors to gas-phase bromine radicals. Ab initio calculations indicate that water stabilizes the BrOOO− intermediate (Figure 5b), but it is not clear how this trend progresses with larger cluster size or how the cluster geometry impacts the energetics. Furthermore, the importance of the water network in the atmospherically significant reaction of O3 with I− is largely unexplored.
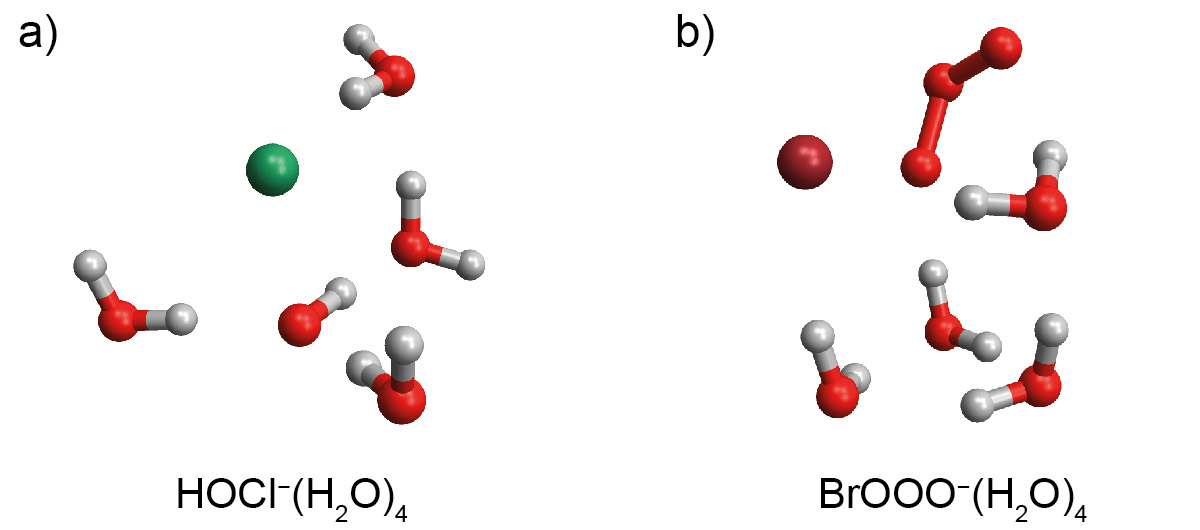
These experiments will provide new insight into the chemistry occurring at the water/air interface. Ab initio simulations and aerosol chamber experiments continue to identify potentially new interfacial reaction pathways, and these studies will provide a connection between these two methodologies by providing experimental characterization at a molecular level. Combining this chemical insight with atmospheric models will advance our understanding of critical issues including urban air quality and climate change.
At the moment, our instrument requires additional modifications to successfully generate the oxidants required to form the clusters of interest, but we are looking forward to working on these projects as our instrumentation progresses.