Bis(phosphine imides)s: Easily Tunable Organic Electron Donors
Vanina G. Guidi, Zhou Jin, Devin Busse, William B. Euler, Brett L. Lucht, Journal of Organic Chemistry, 2005, 70, 7737 – 7743
Abstract
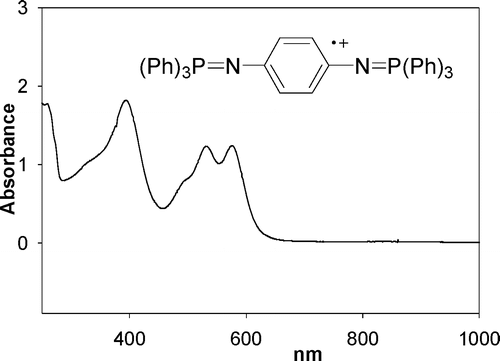
The electrochemical, structural, and spectroscopic properties of bis(phosphine imide)s have been investigated. p-Phenylenebis(phosphine imide)s Ar3PNC6H4NPAr3 (1a-d) have two reversible single-electron oxidations. The first oxidation potentials can be varied from –0.05 to 0.15 V (versus SCE) by modification of the substituents on phosphorus (Ar). Electron-donating substituents lower the oxidation potential, while electron-withdrawing substituents increase the oxidation potential. The difference between the first and second oxidation potential (ΔE, 0.41 – 0.50) and the electronic coupling (Hab, 1.1 eV) are similar for 1a-d. Computational (DFT) and UV-visible-NIR spectroscopic investigations of 1a-d suggest that the first oxidation leads to a delocalized radical 1a•+ while the second oxidation leads to a quinonoidal dicationic state 1a2+. The aromatic linker between phosphine imides has also been modified. Upon oxidation, N,N'-4,4'-biphenylene(bis(triphenyl)phosphine imide) (3) forms radical cationic and a dicationic species similar to 1a-d. While ΔE (0.18 V) and Hab (0.63 eV) are smaller, suggesting weaker electronic communication between the two P=N units in the radical cationic state, the presence of NIR absorptions with vibrational fine structure (768, 861, and 983 nm) supports the formation of delocalized radical cation for 3•+.